最新3D打印患者匹配植入物注册技术审查指导原则要点解读
9月24日,国家药品监督管理局(NMPA)发布了三项骨科植入物注册技术审查指导原则,包括:《3D打印患者匹配下颌骨假体注册技术审查指导原则》、《个性化匹配骨植入物及配套工具医工交互质控审查指导原则》与《生物型股骨柄柄部疲劳性能评价指导原则》。本期,对其中前2项与3D打印骨科植入物密切相关的内容进行要点解读。
3D打印患者匹配下颌骨假体
匹配式人工颞下颌关节产品示意图。来源:中国医疗器械行业协会团体标准《匹配式人工颞下颌关节》
![]() 三种假体
三种假体
《3D打印患者匹配下颌骨假体注册技术审查指导原则》适用于可以实行注册管理的3D打印患者匹配下颌骨假体,全部或部分通过增材制造加工工艺实现,不包括颞下颌关节假体。实行备案管理的定制下颌骨假体也可以参考本指导原则中适用的技术内容。
3D打印患者匹配下颌骨假体用于治疗创伤、良性肿瘤和畸形等多种原因导致的骨骼成熟患者的下颌骨缺损与畸形等。根据假体在体内应力承载方式不同,可分为三类:(1)适用下颌骨节段性缺损并行骨移植修复时,辅助恢复下颌骨连续性、精确固位移植骨段并恢复下颌骨外形,术后早期主要由假体承担应力;或用于在下颌骨骨折或正颌外科术中精确控制并固定各骨段位置(简称“固位假体”);(2)适用下颌骨节段性缺损且未行骨移植修复时,替代缺损区下颌骨组织及功能,完全由假体承担应力,并为最终的功能恢复奠定基础(简称“替代假体”);(3)适用修复下颌骨体积缺陷或轮廓畸形,填充缺损区域(简称“填充假体 ”)。
今年1月1日正式实施的《定制式医疗器械监督管理规定(试行)》中指出,定制式医疗器械与患者匹配型医疗器械均属于个性化医疗器械范畴。两者之间有区别,并且容易混淆。患者匹配医疗器械是指医疗器械生产企业在依据标准规格批量生产医疗器械产品基础上,基于临床需求,按照验证确认的工艺设计和制造的、用于指定患者的个性化医疗器械。患者匹配医疗器械具有以下特点:一是在依据标准规格批量生产医疗器械产品基础上设计生产、匹配患者个性化特点,实质上可以看作标准化产品的特定规格型号;二是其设计生产必须保持在经过验证确认的范围内;三是用于可以进行临床研究的患者人群。如定制式义齿、角膜塑形用硬性透气接触镜、骨科手术导板等。
而定制式医疗器械仅供提出特殊需求出具订单的医疗机构用于指定患者,非订单机构或非指定患者不得使用。
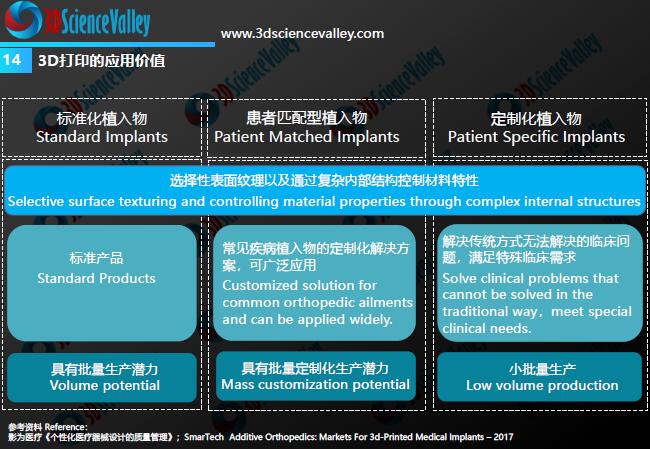
3D打印技术在骨科植入物制造中的应用可以分为:标准化植入物、患者匹配型植入物和定制化植入物。就可广泛应用的患者匹配型植入物而言,3D打印在制造方式上所具有的特殊优势是能够实现不同患者匹配型植入物的批量化生产。
![]() 三种材料
三种材料
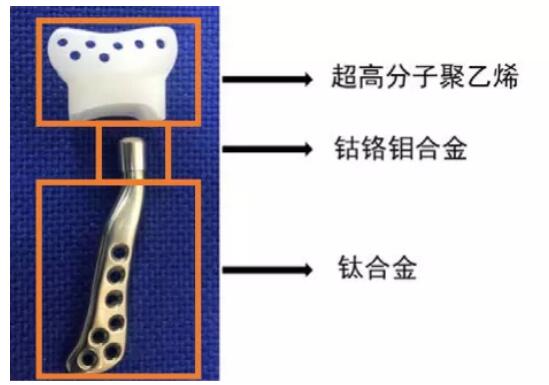
如按照材料将3D打印技术在骨科植入物制造中的应用进行细分,则可以分为金属粉末、聚合物材料(如PEEK)、无机非金属(陶瓷)。这三大类材料3D打印患者匹配下颌骨假体中的应用,指导原则都有所涉及。

指导原则具体指出,针对金属粉末材料,应测定材料的化学成分组成(单一和总和元素限量、氢含量、氧含量等)、纯净度、颗粒形状(圆形度/球形度)、比表面积、粒径及粒度、松装密度、振实密度、流动性等。针对聚合物材料,如PEEK,应测定材料的化学成分组成、线径、密度、热性能、力学性能(如适用)等。针对非金属无机材料,应测定相应的理化指标,例如化学成分组成、平均相对体积密度、微观结构、材料强度、硬度、杨氏模量、疲劳性能等 。
医工交互
《个性化匹配骨植入物及配套工具医工交互质控审查指导原则》适用于个体化下颌骨假体、定制正颌导板及钛板、髋臼周围型肿瘤髋关节假体、骶骨肿瘤假体、个性化匹配多节段人工椎体等个性化匹配产品及配套工具的设计开发。聚焦于“医工交互”的内容,不包括对生产“转换”阶段的讨论,不限于增材制造(3D打印)等具体的生产加工工艺。
![]() 医、工的资质
医、工的资质
该指导原则提出了对主持和参与个性化匹配骨植入物及其配套工具设计制作的医工双方人员的资质要求:“医”方建议由三甲医院中具有正高级职称的医生主持个性化匹配骨植入物及其配套工具的全部流程,该医生团队应有运用传统标准化产品开展同等病患部位与病情下类似手术的既往经验;“工”方参与人员需具有个性化匹配骨植入物设计经验或具有3年以上类似植入术式的非个性化匹配植入物的设计与开发经历。
![]() 设计程序
设计程序
该指导原则归纳了个性化匹配骨科植入物设计以及植入物配套工具设计的主要程序,并明确了每个程序中医生、工程师的职责及交互内容。
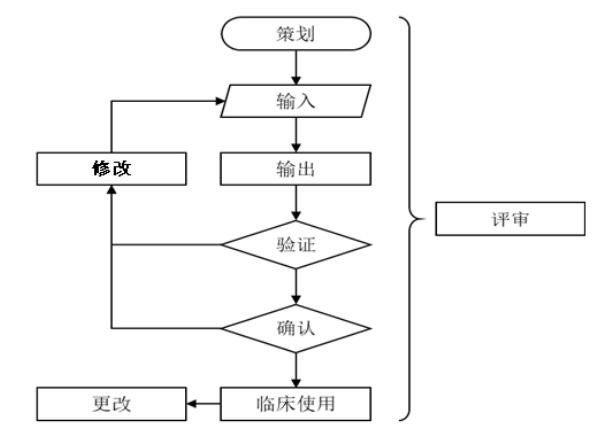
个性化匹配骨植入物的设计程序。来源:《个性化匹配骨植入物及配套工具医工交互质控审查指导原则》
例如:个性化匹配骨科植入物设计的程序包括策划、输入、输出、验证和确认五个主要步骤;在验证不满足要求的情况下,触发修改工作;根据前序已植入产品临床随访结果,对后续的实施病例的产品进行设计更改;设计程序的所有步骤都在评审监督下开展;所有流程都需进行记录与留样。其中,骨植入物的“输出”结果将触发配套工具的设计程序。
![]() 有别于标准化产品的确认与验证
有别于标准化产品的确认与验证
标准化产品需在使用前完成大规模临床研究,而个性化匹配产品仅用于某一特定病患,并不具备大规模临床研究的条件。那么如何对这类个性化产品进行确认与验证呢?
指导原则在验证环节中指出,不同于批量化重复生产的常规标准化产品,个性化匹配骨植入物及配套工具的验证包含了对“个性化加工制造”的验证,故医生必须参与其中,需对每一件个性化匹配骨植入物及其配套工具的设计分别进行。因而,个性化匹配骨植入物的验证包含了“确保其在服役过程中能够安全、有效地替代缺损部位的功能”的目的,对其验证包括设计开发评价、伴随样品的理化性能测试、产品生物力学性能测试等;配套工具的验证需保证其在手术过程中能够准确实现对截骨等关键步骤的辅助定位,其验证主要通过对辅助工具的使用模拟和手术方案验证。
指导原则指出,对于个体化匹配产品,应关注临床确认的抽样统计、代表产品选择、临床评价产品与放行产品的区分。个性化匹配产品的设计确认重度依赖既有类似病例所累积的治疗数据,仿真台架模型及动物模型力学性能测试,而基本上无法在使用前完成大规模临床研究。因此,确认工作应有更灵活、更严谨并动态调整的临床确认方法,将统计学控制贯穿于全生命周期的确认证据,且应基于相近生物力学功能的、同类个体化匹配产品系列的迭代数据。
![]() 有限元分析
有限元分析
有限元分析是在设计开发中评估植入物在人体内的安全性和有效性的重要工具,对于不具备开展大规模临床研究的个性化产品,有限元分析的作用更为突出。
指导原则对于个体化匹配骨植入物及配套工具设计开发有限元分析提出了以下7点注意事项:
1.明确有限元分析的对象、目的及分析结果的评价指标。
2.明确开展有限元分析所使用的软件系统。
3.描述有限元模型的建立依据,需明确指出所建立的有限元模型与实际骨植入物设计模型及其预期使用环境在关键要素上的一致性和差异。
4.个性化匹配植入物的有限元建模。
5.有限元模型需经过验证才能证明其有效性,验证的方式包括常识性判断、与实际物理实验结果对比或与类似产品的研究报告对比等。
6.有限元分析的结果,均应经所设计植入物或工具的动静态力学实测验证,所以应规定测试所需要的样本量。
7.有限元分析的报告应按照预定的格式,按照上述要求叙述建模、验证过程及其合理性,并根据预期评价指标对分析结果进行呈现,呈现形式包括整体、组件和骨骼端的应力/应变分布变化云图、数值曲线等。
![]() 11种植入物的设计流程图
11种植入物的设计流程图
该指导原则给出了11种个性化匹配植入物以及配套的手术规划等工具的设计流程,如:颅、颌面外科植入物、肩关节植入物、胸肋骨植入物、脊柱植入物、骨盆植入物(非髋关节)、膝关节植入物、髋关节植入物、足踝关节植入物、四肢创伤内固定植入物。
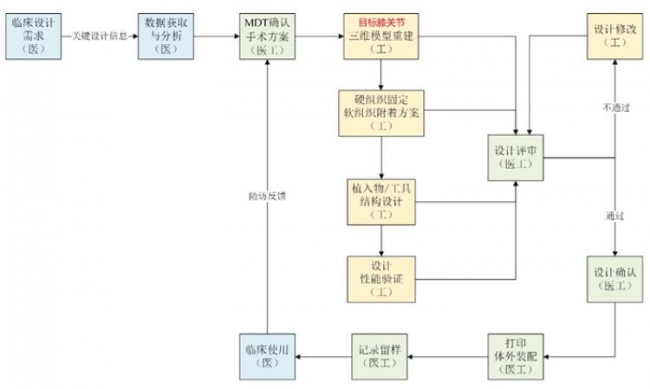
膝关节植入物设计开发医工交互流程。来源:《个性化匹配骨植入物及配套工具医工交互质控审查指导原则》

(责任编辑:admin)

 中南大学:增材制备可生物
中南大学:增材制备可生物 2024年二季度,中国3D打印
2024年二季度,中国3D打印 《Bioactive Materials》
《Bioactive Materials》 增材制造中机器学习研究综
增材制造中机器学习研究综 高性能水凝胶的3D生物打印
高性能水凝胶的3D生物打印 研究人员开创使用X射线和
研究人员开创使用X射线和 选区激光熔化增材
选区激光熔化增材 大型聚合物3D打印
大型聚合物3D打印 6K Additive最新
6K Additive最新 如何打造增材制造
如何打造增材制造 从实验室走向生产
从实验室走向生产 3D打印在口腔修复
3D打印在口腔修复

